
新版TCGA数据转录组表达数据下载及整理(R语言)
简介
由美国05年发起的癌症和肿瘤基因图谱(TCGA,The Cancer Genome Atlas)计划,旨在应用基因组分析技术研究癌症中的基因组变化,做了大规模的基因组测序,样本量过万,包含了三十多种癌症,其中尤其宝贵的是这些样本都有很详细的预后随访信息。TCGA提供了大量的深度测序数据,包括Gene expression, DNA methylation, Copy Number Variant, Mutation还有更深度的exon expression外显子测序结果,最常用的是33种肿瘤及正常组织的高通量芯片或测序数据,其次包括10种罕见肿瘤,无疑是一座巨大宝库。此外其临床数据包含。
TCGA数据下载网址:https://www.cancer.gov/ccg/research/genome-sequencing/tcga
TCGA数据概况
-
Clinical: 包括病人的一般情况、诊治情况、TNM分期、肿瘤病理、生存情况等。
-
mRNA表达数据: 通过mRNA芯片或者RNAseq测得的mRNA表达量
-
microRNA: microRNA芯片或者microRNA-Seq测得的microRNA表达量
-
Copy number variation: SNP芯片得到的肿瘤组织比对正常组织的染色体上各片段的比值
-
Mutation: 肿瘤组织测序结果相对参考基因组的核苷酸突变,包括插入和缺失等变化
-
Protein: 蛋白芯片测序得到的约200种常见癌症相关蛋白的表达量Mythelation: 甲基化芯片测得的DNA甲基化数据,主要为27和450两种芯片的数
转录组数据的下载
进入网页:
https://www.cancer.gov/ccg/research/genome-sequencing/tcga
①选择Access TCGA Data
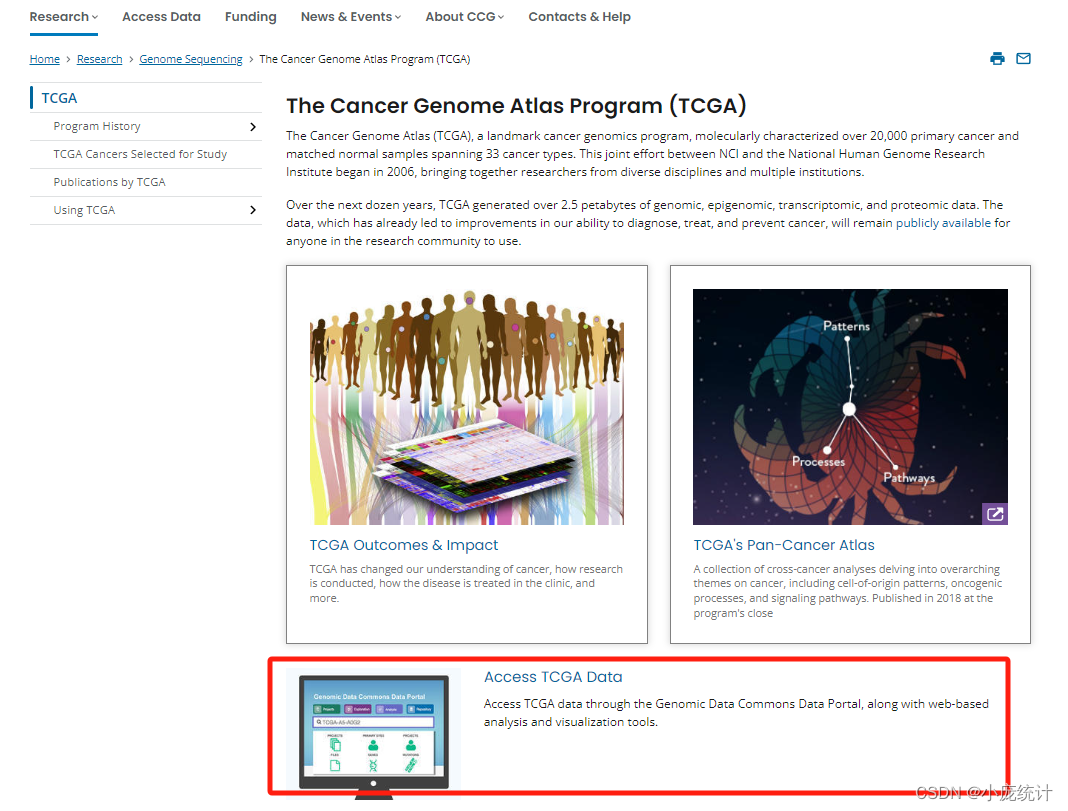
②选择Projects
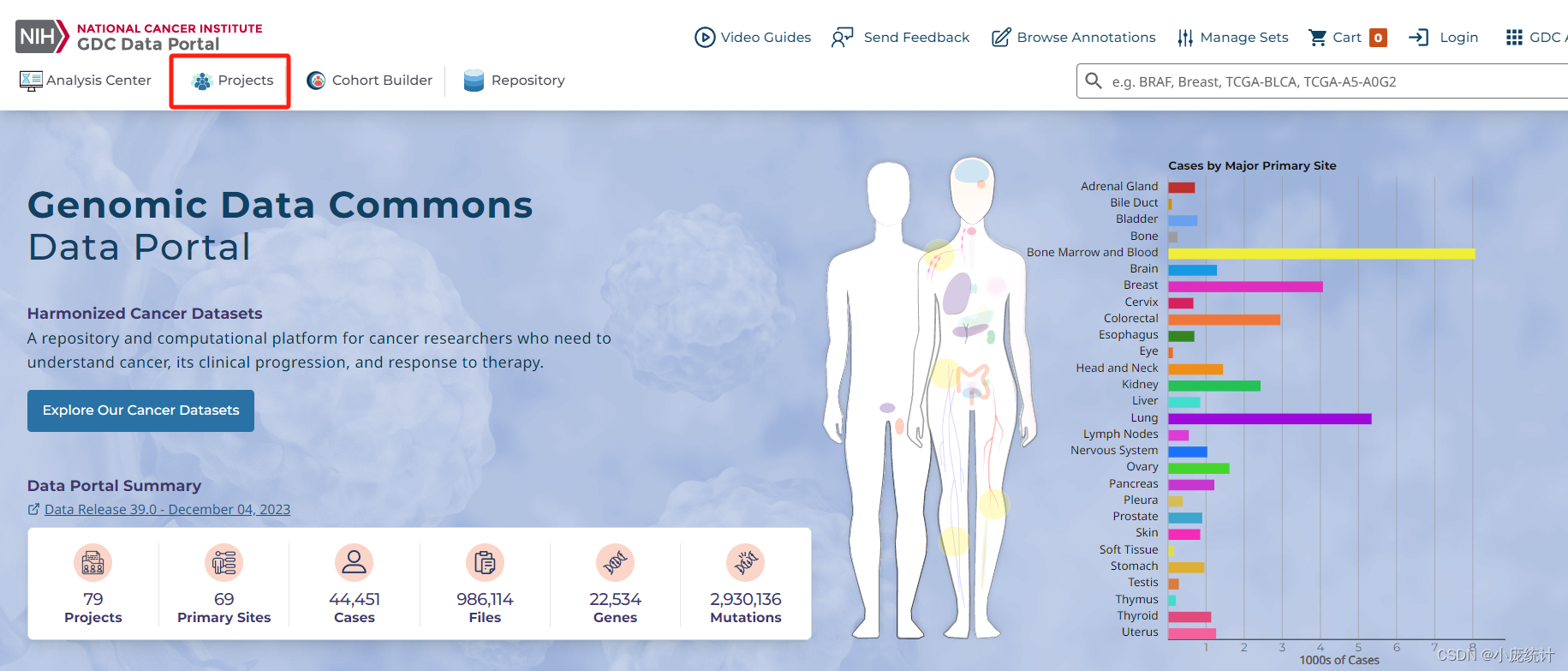
③左侧的原发部位Primary Site,选在自己的方向(以乳腺癌为例)
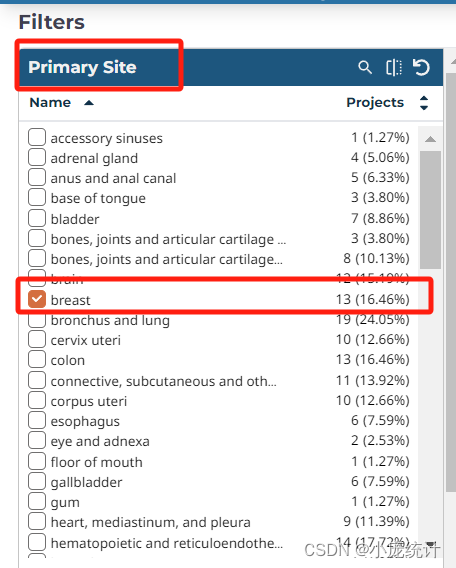
④项目Program选择TCGA

⑤选择队列Cohort Bulider
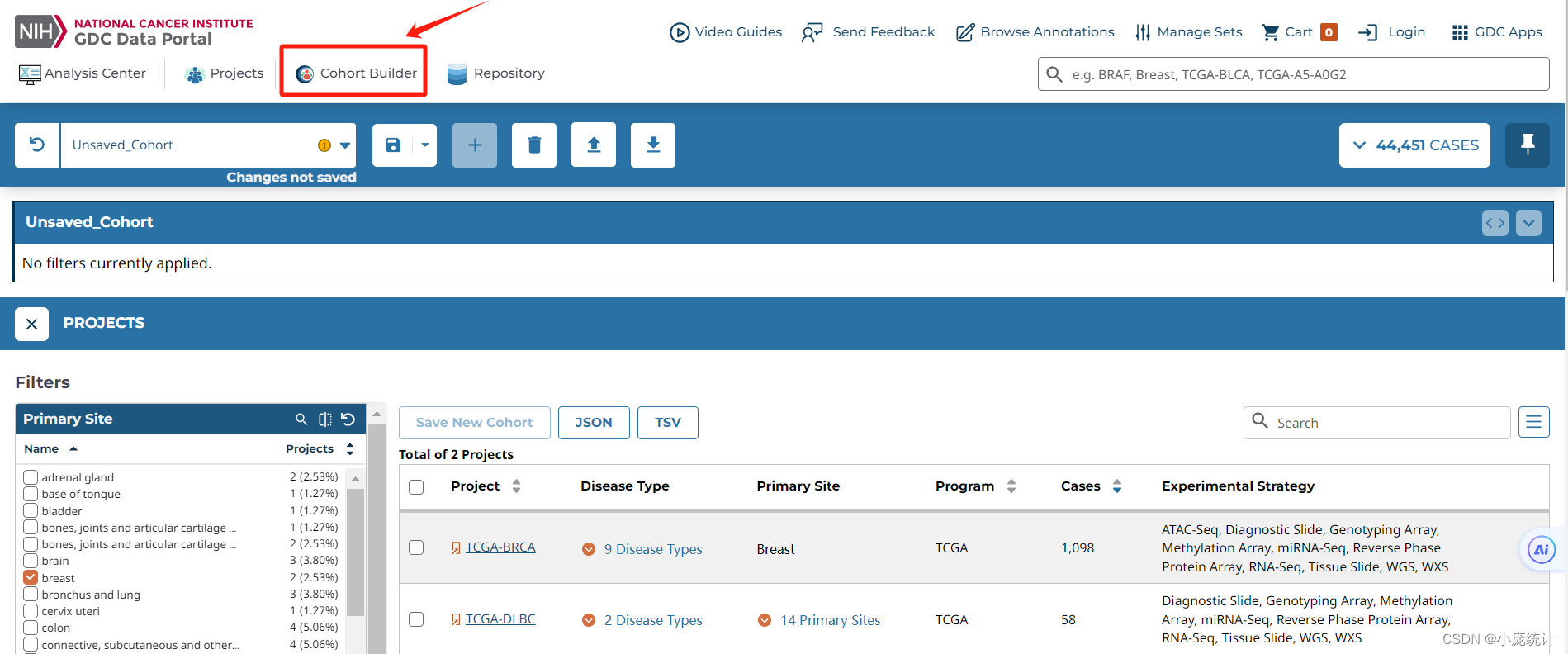
⑥在Program栏目选择TCGA
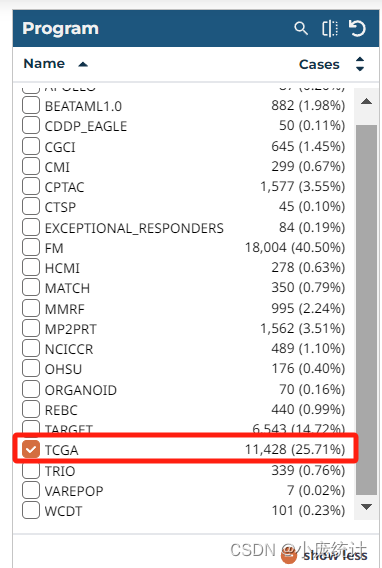
⑦在Project栏目选择简称TCGA-BRCA

⑧选择Repository
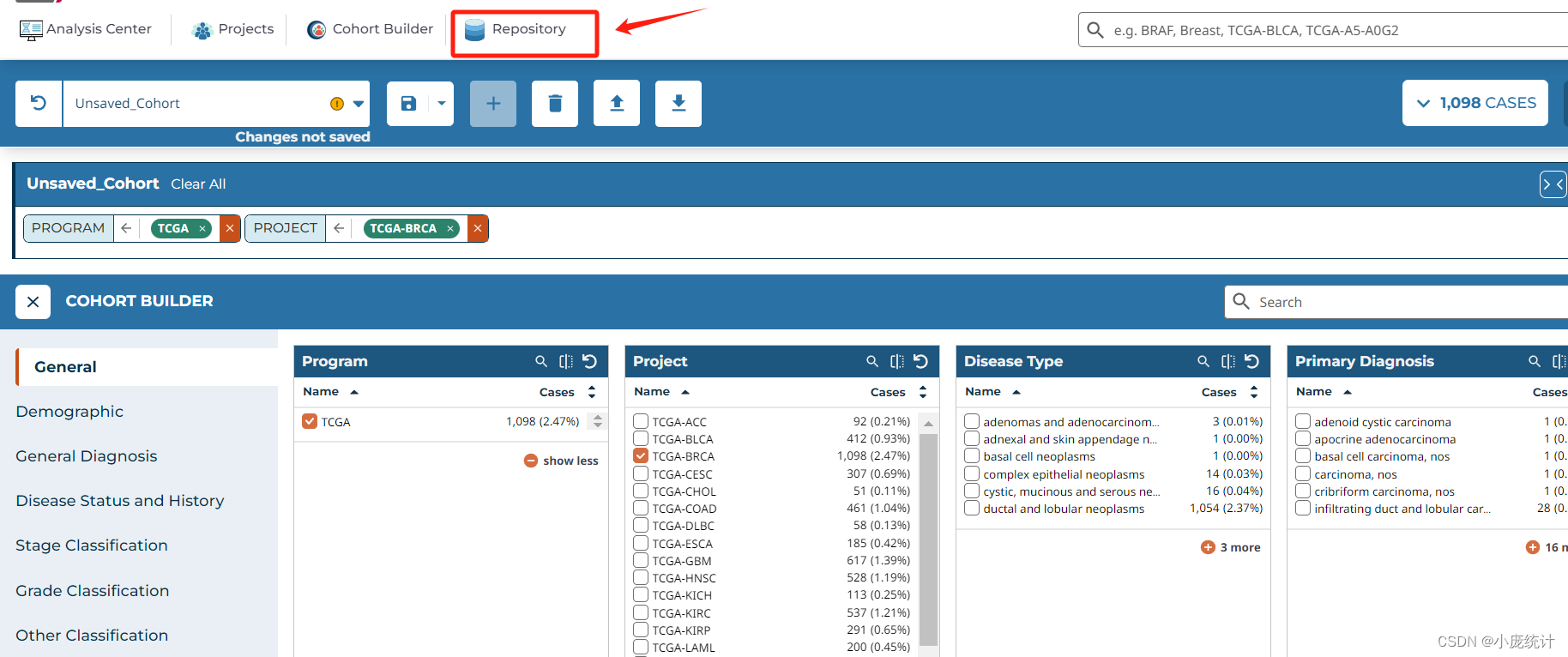
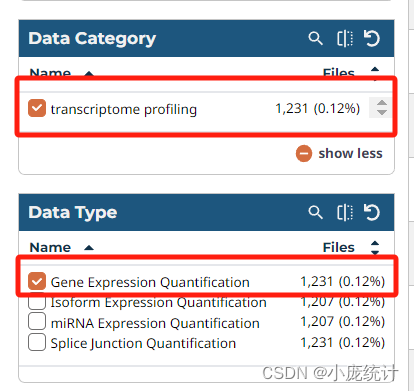
⑨在左侧找到Data Category选择转录组数据Transcriptome profiliing
数据类型Data Type选择Gene Expression Quantification
⑩加入到cart
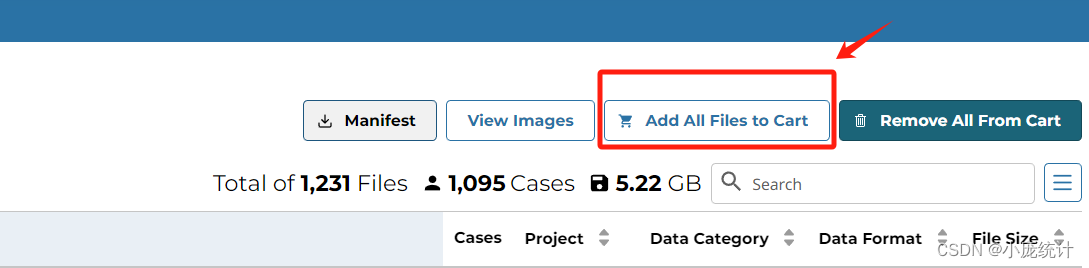
下载两个文件分别是Cart文件和Metadata文件
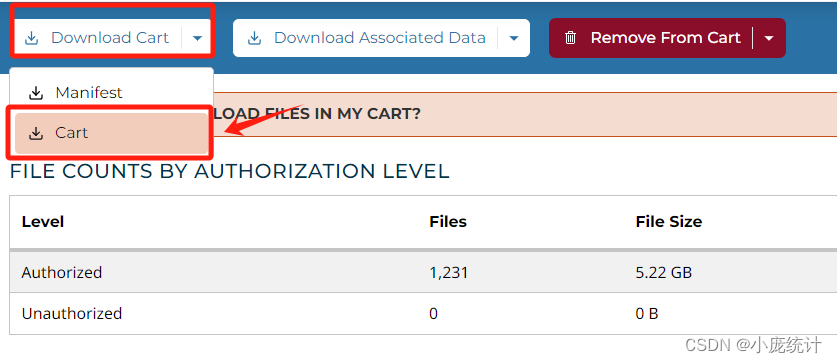
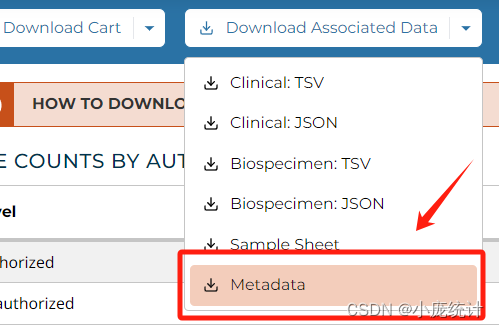
下载完成后我们将Cart,解压在他的原始文件夹中,我们可以打开其中一例数据看看包含哪些项目

包含项目:
-
gene_id:此处的为ENSMBLE格式;
-
gene_name:symbol格式
-
unstrandes:基因的表达counts值
-
tpm_unstranded:TPM值
-
fpkm_unstranded:FPKM值
mRNA-Seq数据分为4种:
Counts;TPM;FPKM;FPKM-UQ。其中Counts属于原始的格式

Counts: 测序的reads中比对到某个基因上的计数;TPM、FPKM: 用来衡量转录本表达丰度的一种量度方式;UQ-FPKM:通过上四分位点进行标准后的FPKM;
数据格式转换参考:
https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
使用R语言数据整理FPKM数据
library(rjson) library(limma) setwd("C:\\Users\\TCGA-BRCA") #此处我将下载的数据,均放在TCGA-BRCA文件夹中,更改为自己的文件夹 metafile="metadata.cart.2024-03-24.json" #下载的metadata文件的名称 gdcfliename="gdc_download_20240324_144347.209765" #cart文件的名称 path1="gdc_download_20240324_144347.209765\\" #cart文件名+“\\” outfilename="TCGA-STAD_FPKM.txt" #输出表达矩阵文件的名称 #为了方便大家使用,大家只用修改以上内容 json = jsonlite::fromJSON(metafile) id = json$associated_entities[[1]][,1] sample_id = sapply(json$associated_entities,function(x){x[,1]}) file_sample = data.frame(sample_id,file_name=json$file_name) count_file
-



